Change Control Management Procedure

1.0 PURPOSE:
To ensure that any change related to quality system, manufacturing, testing, and distribution are reviewed, assessed, and implemented only after evaluation, approval, and documentation.
2.0 SCOPE:
This procedure applies to the introduction and change/s in
Facility / Equipment / Instrument / Support system
Product Quality / Process / Labelling / Material / Analytical methods / Specifications
Changes in System / Procedure / Document / Computerized Systems / Software / Hardware
Any other element that can impact product quality or safety.
3.0 RESPONSIBILITIES:
3.1 Initiator
3.2 Department Head / Designee
3.3 Quality Assurance - Reviewer
3.4 Change Control Committee
3.5 Head Regulatory Affairs / Designee
3.6 Head QA/ Designee
4.0 DEFINITIONS:
4.1 Change: Any addition to, deletion of, or modification to manufacturing facility, utilities, process, material, product, procedures, or equipment (including software) that impacts quality or regulatory requirements.
4.2 Changes can be classified as follows:
Minor
Major
Critical
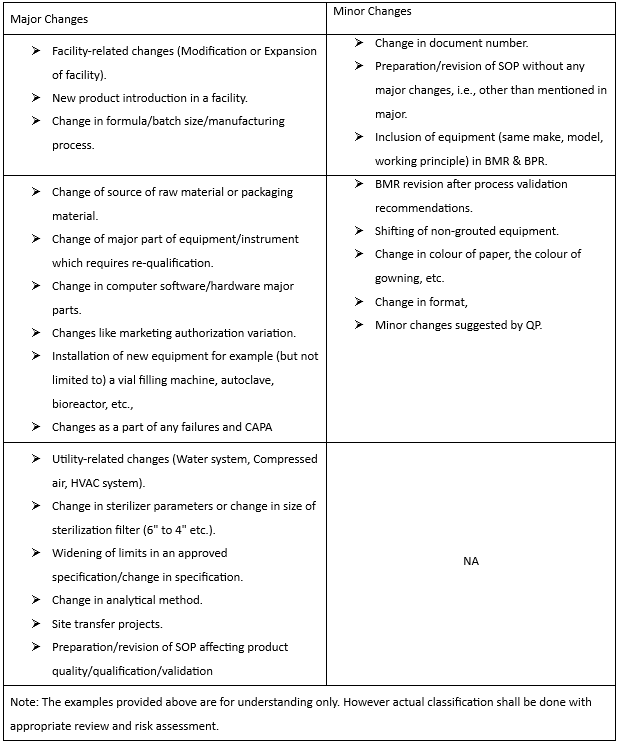
4.3 Minor Change: A change that does not have any impact on the identity, quality, purity, strength, stability, safety efficacy, or physical characteristics of the product.
4.4 Major Change: A change that may have an impact on the identity, quality, purity, strength, stability, safety, efficacy or physical characteristics, or regulatory filing of the product.
4.5 Critical Change: A change that has a significant impact on the identity, quality, purity, strength, stability, safety, and efficacy or physical characteristics of the product.
5.0 PROCEDURE:
5.1 A change to the following shall be subjected to the Change Control Form but not limited to,
5.1.1 New change control, concerning the facility, equipment, application software, utility, document, product, and process.
5.1.2 Changes in Facility
5.1.2.1 Facility Design/Facility Layout/Engineering Drawings.
5.1.2.2 Change in Location or Addition/Alteration and discontinuation of manufacturing site.
5.1.2.3 Change in HVAC Parameter
5.1.3 Changes in Equipment / Changes in Software
5.1.3.1 Major Changes in the Existing Equipment / Instruments.
5.1.3.2 Introduction of New Equipment / Instruments.
5.1.3.3 Calibration tolerances (or) Frequency of calibration.
5.1.3.4 Critical utilities such as water for injection plants, Pure steam, filtered air supplies, power, and vacuum systems.
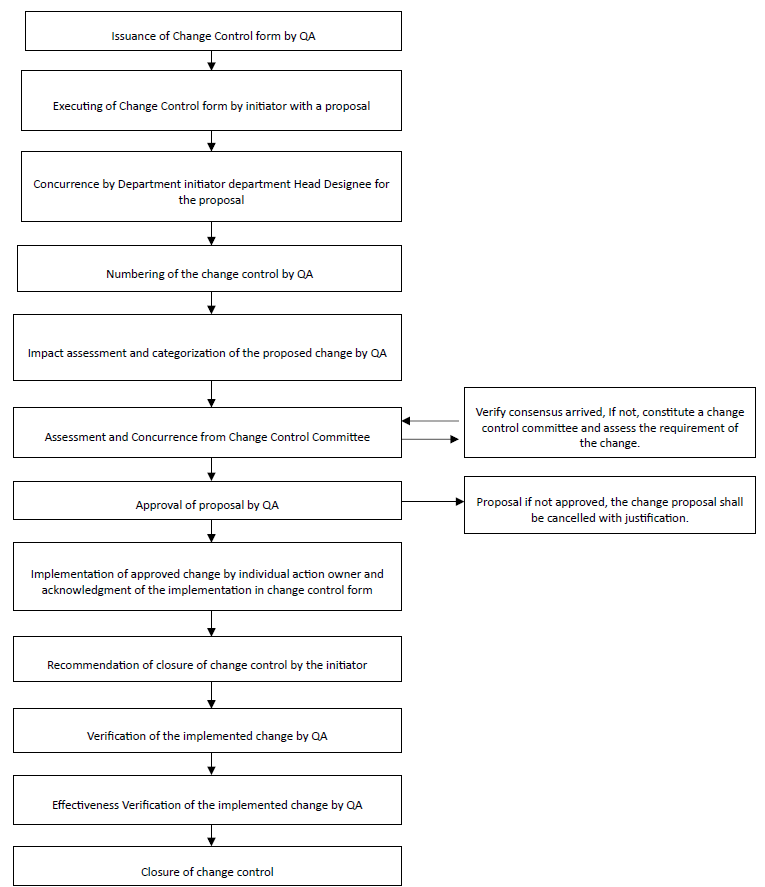
Note: For the Flow chart for Change Control Procedure refer the Annexure QAP/GEN/ANX/XXX-0001
5.1.3.5 Any change in the frequency of preventive maintenance or calibration.
5.1.3.6 Transfer of equipment from one location to another location.
5.1.3.7 Change Control Form is not required in case of portable equipment and weighing Balance transfer from one location to another location within the department.
5.1.3.8 If any gradation of patches or service packs then change Control Form shall be raised.
5.1.4 Changes in Process/Product
5.1.4.1 Addition/Deletion/Modification of RM/PM (or) quantity in manufacturing/ packing process.
5.1.4.2 Any changes in the Manufacturing process (Method of manufacturing).
5.1.4.3 Any change with respect to product in Batch size and storage conditions.
5.1.4.4 Any change in the cleaning method and cleaning procedures.
5.1.4.5 Any changes made after process validation/cleaning validation.
5.1.4.6 Any new vendor or alternate vendor or any new material from the existing vendor introduced.
5.1.4.7 Any changes in materials, vendor, and specifications as per regulatory requirements.
5.1.5 Changes in Materials
Any change in Raw materials and or change in grade or pH of material.
Any change in Packing Materials.
Any changes in the quantity of material
5.1.6 Changes in Testing (Material and Finished Product)
Any changes in analytical method validation.
Any changes required in specifications due to changes in Pharmacopoeia.
Product/Material Specifications (Addition/Deletion of any tests).
Any changes in the Method of Analysis.
Any changes in Stability testing and Stability Protocols.
Any changes made in the specifications due to trend/Annual Product Quality Review.
Any changes due to planned deviations as a part of Continuous improvement.
Any changes in the Shelf-life of the product.
5.1.7 Changes in Documents
Any changes as per cGMP regulations
Any changes in Master formula / Batch Manufacturing Documents/Master Documents
Any changes made in Quality systems SOP's/STP's/General Test Procedures/ Specifications, Quality Agreements.
Any changes in In- process /Finished product Labels.
Revision of existing documents.
5.1.8 In case of any document to be made obsolete, the Change Control Form shall be raised along with justification/reason.
5.1.9 In case of shifting of any SOP from one department to another department.
5.1.10 This procedure shall provide a formal change request form system to prevent unauthorized and unreported changes to established facility systems and processes.
5.2 In case of any miscellaneous study protocol, effectiveness check protocol, etc., any QMS document (i.e., CAPA, Incident, Deviation, OOL, etc.) number can be referred. If no QMS document is available, then change control shall be initiated.
5.3 Change Control Form Initiation:
Note: The procedure applies to both temporary and permanent change control.
5.3.1 The change proposal will be categorized as Minor or Major depending on the nature and degree of changes and product quality impact based on the below table.
5.4 Timeline for change control:
5.4.1 For temporary change control: The duration shall be confined to the activity period of change or number of batches or any other particulars to be recorded in the change control form.
5.4.2 For permanent change control:
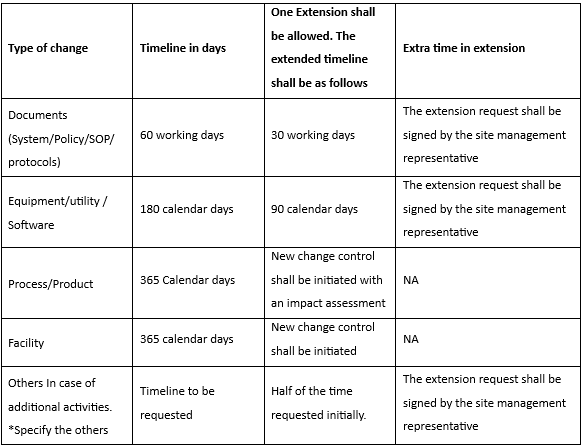
5.4.3 The change proposal shall be approved within 30 working days from the change control form numbering.
5.4.4 If the change is not approved within 30 working days, the proposed change shall be considered cancelled.
5.4.5 If the change is not implemented even after the extended date the same shall be cancelled with impact assessment and interim control as applicable by initiator HOD/designee. If the change control is partially implemented the same shall be closed with impact assessment and interim control as applicable by the initiator HOD/designee, if required new change control shall be initiated to address the unexecuted action points same.
5.4.6 In case change control form remains open for more than 12 months from the date of initiation, Head- QA in consultation with change owner can close the change request form in "AS IS" status, unless the activity (is) in progress. To implement such changes again they shall be routed through new change control form. Head QA shall comment the same in the disposition status.
6.0 REFERENCE:
6.1 ICH Q10: Pharmaceutical Quality System
6.2 EU GMP Guidelines Annex 15
6.3 Procedure for preparation of standard operating procedure
6.4 Document Control
6.5 21 CFR Part 201 & 211
7.0 ABBREVIATIONS:
7.1 QA- Quality Assurance
7.2 RM-Raw Material
7.3 PM-Packing Material
7.4 cGMP-Current Good Manufacturing Practices
7.5 SOP-Standard Operating Procedure
7.6 STP-Standard Test Procedure
7.7 BMR-Batch Manufacturing Record
7.8 BPR-Batch Packing Record
7.9 WFI-Water for injection
7.10 SME-Subject Matter Expert
7.11 SISPQE- Strength, identity, safety, purity, quality, and efficacy
7.12 HVAC- Heating ventilation and air conditioning systems
7.13 QP- Quality person
Annexure-1
FLOW CHART FOR CHANGE CONTROL
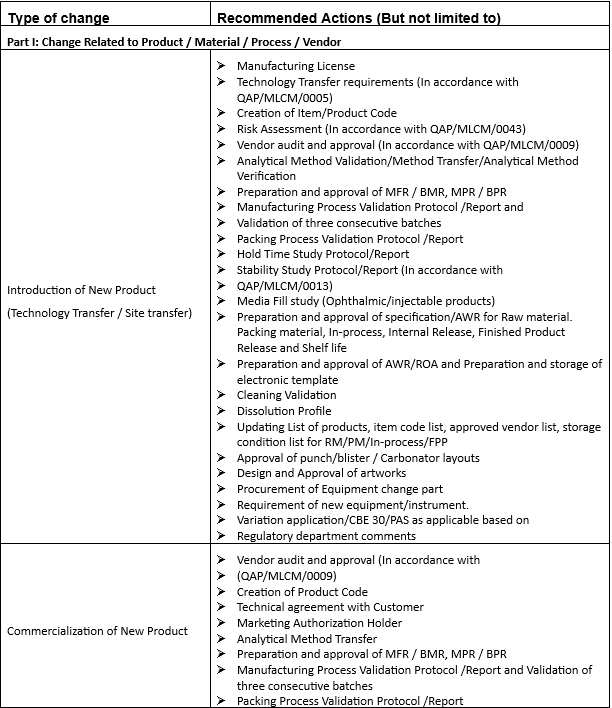
Annexure-2
GUIDANCE FOR IMPACT ASSESSMENT
Get ready to use editable documents in MS Word Format (Regulatory standard SOPs with Formats,Templates and Annexure) Only Rs-99₹/-
Contact:
Email: contact@likeways.co.in
WhatsApp/Telegram No: 9738137533 (Only for Message)
Note: Kindly note that you will receive the document through email only after the payment has been completed. Please send the screenshot of your payment confirmation via WhatsApp/Telegram. A payment QR code will be provided from our end.